4 Input Data
4.1 File Format
Hourglass currently supports input data as csv (comma separated values) and txt (tab-delimited text) files
Requirements
- All files should have row and column names.
- Missing values should be denoted NA or left blank (not nan, N/A, “NA”)
- Avoid special characters, such as /,\,+,-,^ in column names. Use descriptive names with underscores (i.e. _) or periods (.).
4.2 The 3 Files
Your dataset comprises of 3 tables or files:
The file descriptors are named below with abbreviations in brackets. For R package users, these are names of elements in the dataset (ds) list object.
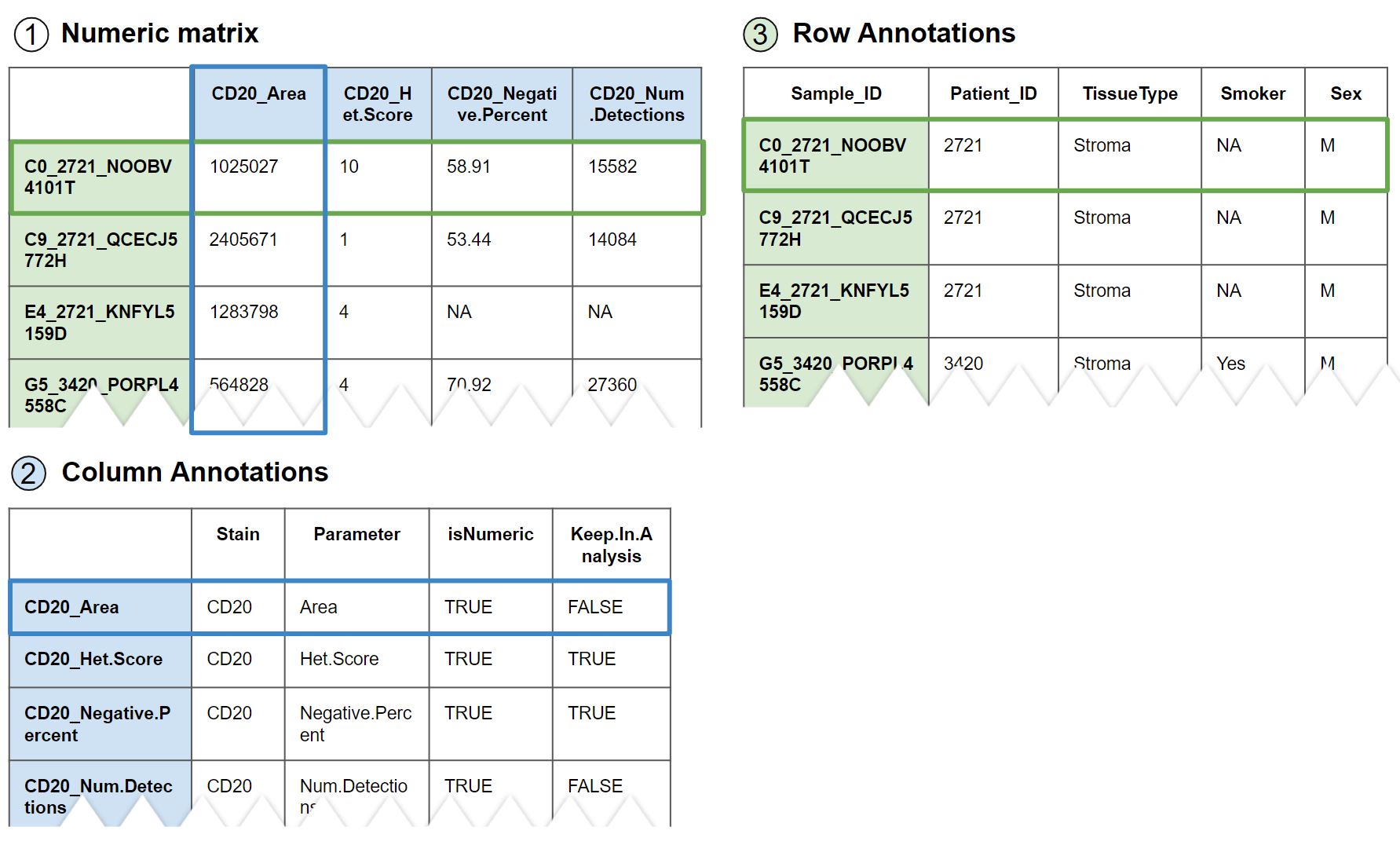
Figure 4.1: Snippet of the three files/tables required for Hourglass: 1) values matrix, 2) metadata/annotations for columns of values matrix, and 3) metadata/annotations for rows of values matrix.
1. Numeric matrix (referred to as ‘numMat’/‘vals’)
= Numeric values from quantification, where rows (samples/patients) x columns (genes/features/parameters). For best output results, columns should contain feature first name with a period to delimit the rest of the column name. (e.g. try “Gene1.Parameter”, not “Gene1_Parameter”); for heatmaps/correlation plots, the part of the column name after the period will be removed by Hourglass.
For example, for RNA sequencing data: data matrix = expression matrix, columns = genes, rows = samples.
2. Column Annotations (or ‘colAnn’)
Annotations/descriptors for columns in numMat. At least two columns are needed for “Feature” and “Parameter” (can be called something else).
= Feature/parameter information, describes the columns of numMat. First column must be the column names of numMat.
- Descriptive columns names in data (recommended), e.g. feature_parameter
- For example, for the column “IL6.Number.of.Positive.Pixels”, the feature is IL6 and parameter is Number.of.Positive.Pixels. For the column “Gene1_woo_ahaha_Area”, the feature is Gene1 and parameter is Area, which will be reflected in the colAnn columns.
- Hourglass also subsets columns based on a column called “Keep.In.Analysis” - TRUE to keep, FALSE to discard.
3. Row Annotations (or ‘rowAnn’)
Annotations/descriptors for patient/sample in numMat
= Sample/patient information, describes the rows of numMat. First column must be the row names of numMat.
These are labels or groups that will define main comparisons or groups to compare or for exclusion/inclusion of patients/samples (e.g. removing smokers)
Examples
- For samples:
- Tissue type
- Sample_Cancer_Subtype
- For patients:
- Patient_Cancer_Subtype
- Smoker
- Age at diagnosis
A column for “Patient_ID” is required (can be called something else) for paired analysis (i.e. averaging samples from the same patient) and patient level analysis (i.e. averaging samples from the same patient).
Note: this is external information from clinical charts / prior analysis that is integrated manually prior to running Hourglass. This means transcriptomic, genomic, and clinical calls are created independently of Hourglass and merged to the master rowAnn table via sample or patient identifiers.
4.3 Similar Format
The data input was inspired by popular bioinformatics packages, with numeric data plus metadata.
- Struct is a package in which a simple DatasetExperiment object contains sample measurements, sample metadata, variable metadata. Figure 1 here.
- SingleCellExperiment object contains primary and transformed data (cell x feature matrix) and reads in feature and cell metadata. This is similar to Hourglass running the original and imputed versions in the same run. Figure 4.1 here.
- SummarizedExperiment object similarly contains multiple assays (feature by sample matrices) with colData and rowData. Section 2 here.
4.4 Example Dataset
We included an example multiparametric dataset with annotations for patients/samples. This is an entirely fake dataset.
Its purpose is to showcase the functionalities of Hourglass and help users recreate plots seen here. These values have no biological relevance or connection to literature. Parameters and annotation labels were inspired by the quantified immunohistochemistry dataset for pancreatic cancer tissue microarray.
Features of example dataset
- 30 patients with 1-6 samples per patient
- 102 samples in total (rows)
Features
The following stains (referred to as “features” see Terminology) were quantified:
- CD3, CD8 (which are T Cell markers)
- CD20, CD27, CD5, PDL1 (B Cell markers)
- IL6, SMA (other)
Parameters
The parameters measured for each feature are:
- Area = area of tissue image in mm^2 (random)
- Num_Detections = number of detected pixels on tissue area = Num_Positive + Num_Negative
- Num_Negative = number of pixels of negative staining (random)
- Num_Positive = number of pixels of positive staining (based on trends), affects the following:
- Positive_Percent = Num_Positive / Num_Detections x 100
- Negative_Percent = 100 - Positive_Percent
- Pos_Pixel_Percent = Num_Positive/10^3 x random value from 0.3 to 1.5
- Num_Pos_per_mm_2 = Num_Positive / Area x 10^4
Possible Feature Sets
- TCell = CD3, CD8
- BCell = CD20, CD27, CD5, PDL1
- Immune = TCell, BCell
- All = Immune, IL6, SMA
Possible Feature Parameters
- Num_Pos_per_mm_2
- Pos_Pixel_Percent
Summary of Trends
Most values differ/segregate based on 3 sample cancer subtypes (A, B and C). When there are more than 2 samples per patient, there is a predominant “Patient Cancer Subtype” but some samples might deviate from this (ie. intrapatient heterogeneity).
The following trends are:
- Overall survival is low for A, medium for B, high for C
- Average values for TCell (overall low) vs. BCell (overall high)
- Only for TCells:
- Het.Score low for A, medium for B, high for C
- Smokers for all subtypes for TCell stains are high
- Only for BCells:
- NeoAdjuvant values are lower
- IL6 different between A,B,C subtypes only seen in females (“Sex==F”)
- No trend in SMA
- Random outliers (very high value, 4 per row) and random NAs (8 per row)
How to Access Files
Download 3 csv files here, file names:
- “Example_IHC_sample_colAnn.csv” (colAnn)
- “Example_IHC_sample_rowAnn.csv” (rowAnn)
- “Example_IHC_sample_vals.csv” (numMat)
A corresponding example analysis that is the output of the desktop application, serves as the log file for user options and serves input into the R side (See Output: Excel File can downloaded found here).
How to Access in R
See Example Dataset in R.